



AG Computational Metagenomics
Über 99% der in der Natur beobachteten mikrobiellen Spezies sind nicht kultivierbar, was sie für klassische genomische Studien unzugänglich macht. Metagenomik und Einzelzell-Genomik sind zwei Ansätze, um die "mikrobielle dunkle Materie" zu studieren.
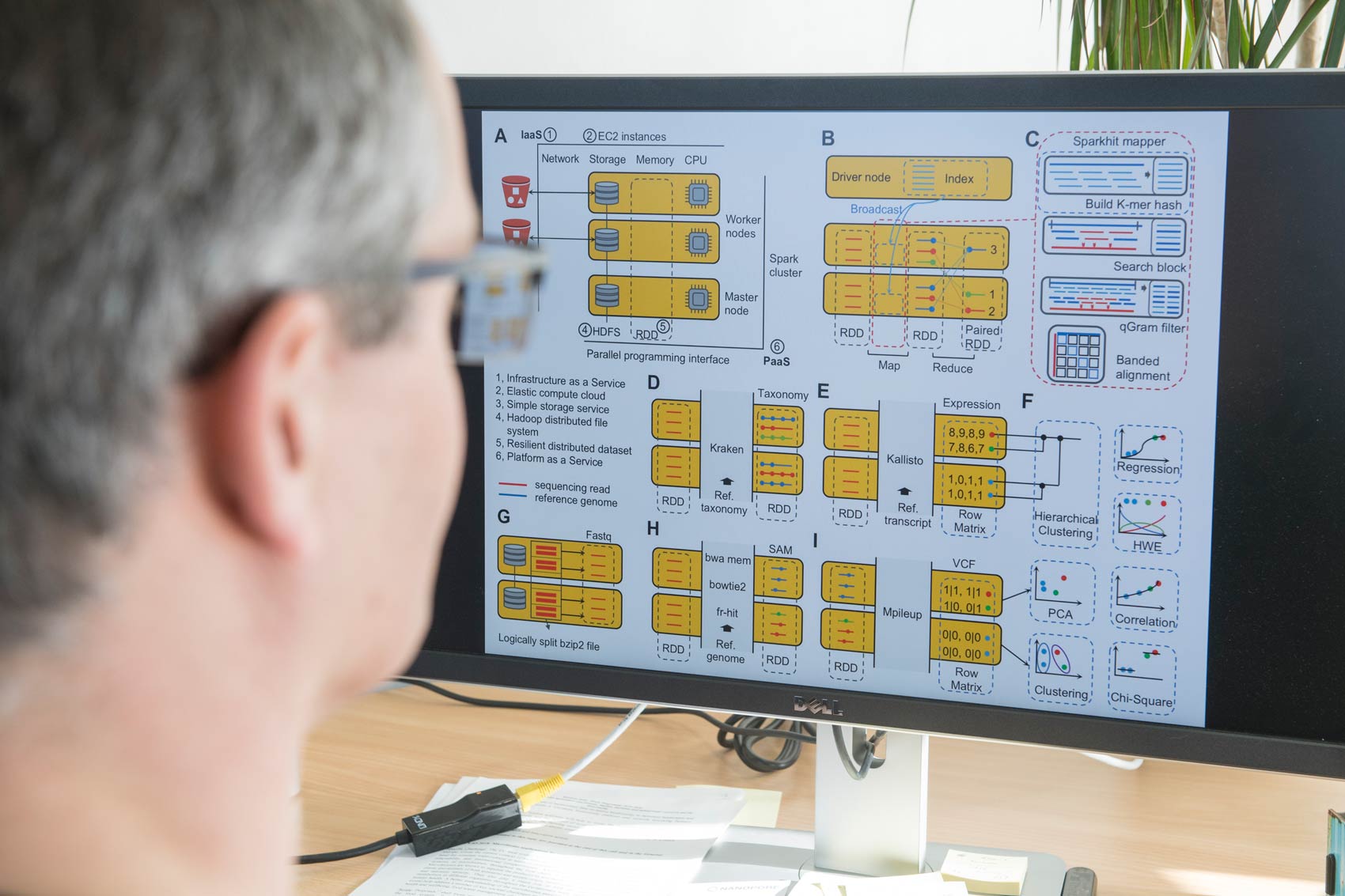
Metagenomik, die direkte Analyse von aus Umweltproben gewonnenen DNA, stellt eine Strategie dar, um die Vielfalt der mikrobiellen Welt zu enthüllen. Aktuelle Sequenztechnologien können in einem einzigen Experiment mehr als ein Terabyte Sequenzdaten erzeugen, was eine sequenzbasierte metagenomische Rekonstruktion vollständiger Gene oder sogar ganzer Genome aus Umweltproben ermöglicht. Die Arbeitsgruppe Computational Metagenomics, geleitet von Prof. Dr. Alexander Sczyrba entwickelt Bioinformatik-Tools und Pipelines für die Analyse von Metagenomik-Studien. Da die Datensätze extrem schnell wachsen, ist ein besonderer Schwerpunkt unserer Forschung die Anwendung von Cloud-Computing-Technologien, um die Analysen auf großen Rechenressourcen dynamisch zu skalieren.
Ein komplementärer Ansatz zur Sequenzierung der DNA einer ganzen mikrobiellen Gemeinschaft ist die Einzelzellgenomik. DNA-Sequenzierung aus dem amplifizierten Genom einzelner Zellen erlaubt es, die Genome von unkultivierten Spezies zu untersuchen. Allerdings stellt das enorme Bias, das durch die Amplifikation und mögliche Probenkontamination entsteht, eine Herausforderung für die Bioinformatik-Analyse dar. Die Forschung in der Gruppe konzentriert sich derzeit auf bioinformatische Ansätze zur Vorverarbeitung von Einzelzell-Sequenzdaten und automatisierten Erkennung möglicher Probenkontamination, eine schwierige Aufgabe, wenn das Zielgenom nicht eng mit einem zuvor sequenzierten Genom verwandt ist.
Ein vielversprechender Ansatz für künftige metagenomische Studien ist die Kombination von Hochdurchsatz-Metagenom-Sequenzierung und groß angelegter Einzelzell-Genomik. Beide Datenquellen können zu Bioinformatik-Analysen kombiniert werden, um ein besseres Verständnis der phylogenetischen Vielfalt der mikrobiellen Gemeinschaften, ihrer Bevölkerungsstruktur und ihrer Funktionalität zu gewinnen.