Bielefeld Institute for Bioinformatics Infrastructure




EU SIMBA project
Analyzing large scale metagenomics data on the de.NBI Cloud


Liren Huang; Bielefeld University, Bielefeld
SIMBA (Sustainable Innovation of Microbiome Applications in the Food System) is a European innovation project, funded under the EU’s Horizon 2020 Funding Programme, which provides a holistic and innovative approach to the development of microbial solutions to increase food and nutrition security.
SIMBA focuses in particular on the identification of viable land and aquatic microbiomes that can assist in the sustainability of European agro- and aquaculture. Under the scope of the EU
Simba project, our research group focuses on exploring microbial communities in large scale publicly available environmental sequencing (metagenomics) data and association studies with plant growth promoting bacteria (PGPB). To that end, our study addresses both computational intensive challenges of searching hundreds of terabytes of public data and sophisticated data mining (e.g. network analysis) on putative PGPB genomes.
We established a scalable bioinformatics workflow for detecting PGPB associated microbes from public data. In particular, we have developed a distributable framework, Sparkhit, that enables screening and mapping [1] terabytes of sequencing data within hours. After preliminary screening of large datasets, EMGB is used as a general purpose bioinformatics workflow for analyzing metagenomics data and visualizing annotation results. We also developed a de-replication tool that can handle large amounts of metagenomics samples and facilitate downstream co-occurrence network analysis. Most tools are containerized (e.g. Docker) and are easily accessible on the cloud.
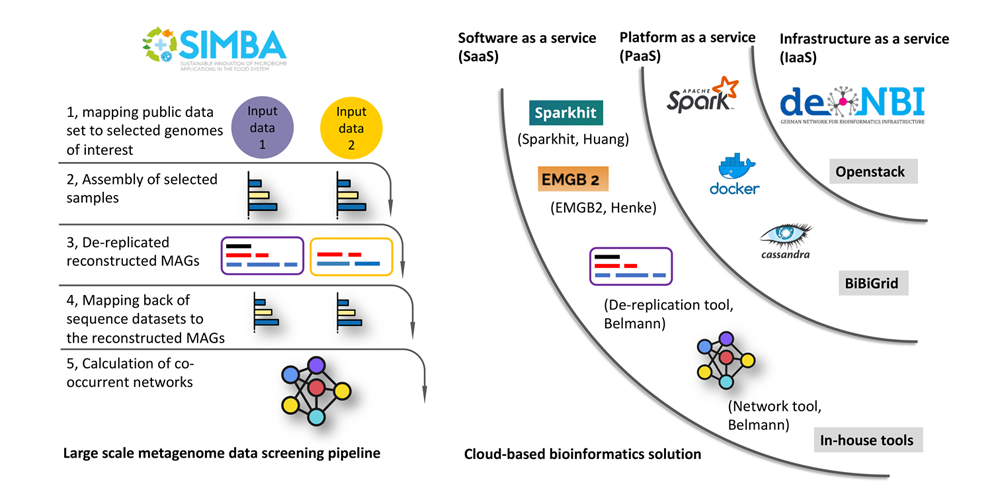
Large scale metagenome data screening pipeline (left) and cloud-based bioinformatics solution (right).
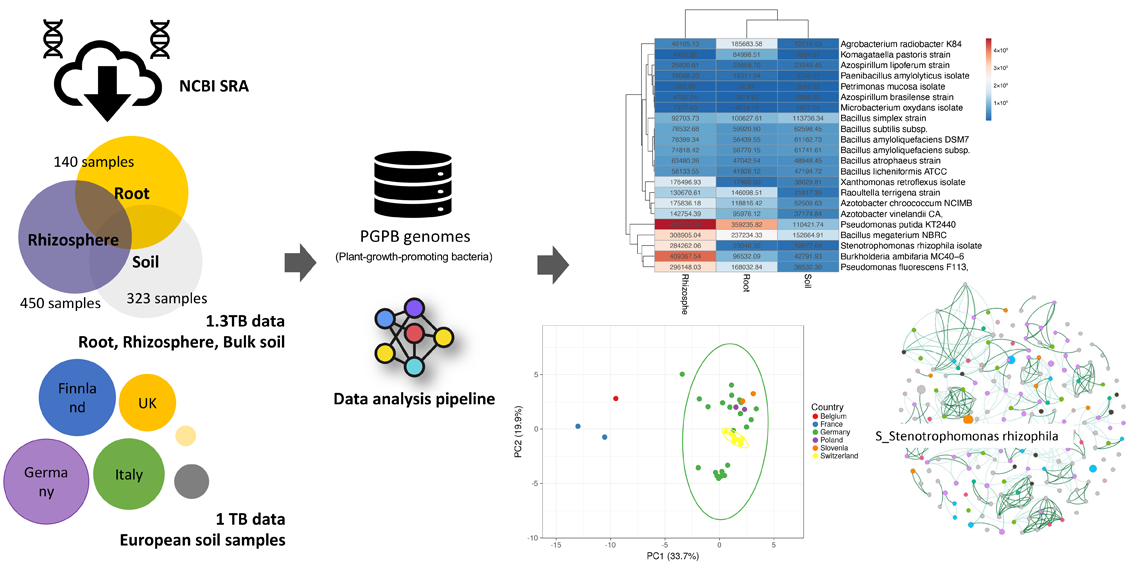
In the case of the EU Simba project, terabytes of public soil metagenome datasets were collected and downloaded on the de.NBI cloud object storage. Associated metadata containing detailed description of the datasets was categorised. In the first step of our analysis pipeline, Sparkhit is used to map all input sequencing data to the selected PGPB genomes. Sparkhit is an in-house fragment recruitment tool that can be scaled to hundreds of computer nodes. Once high similarity hits are found, corresponding samples are selected for assemblies or co-assemblies (multiple samples in one bio-project) using the EMGB pipeline. The EMGB pipeline also generates “metagenome assembled genomes” (MAGs) after assembly, representing the microbes present in the samples.
To remove redundancy between different samples, the generated MAGs are de-replicated and the representative MAGs are selected for further analysis. To refine our analytical pipeline, we have combined a set of existing tools for the de-replication of MAGs. By comparing and evaluating these tools, we were able to identify the best approach to de-replicate our reconstructed MAGs, and accordingly established a personalized de-replication pipeline.
Our de-replication pipeline starts by filtering MAGs with high contaminations and low coverages. After the filtering step, Average Nucleotide Identity (ANI) methods are applied to determine species and strain level clusters. Once clusters are formed, representative MAGs are selected based on the ranking algorithm to represent each cluster. Since the core task of MAG dereplication workflows is the estimation of similarity between genomes, which can be done by calculating ANI, we collected and evaluated several ANI-based approaches. Three different datasets (unfiltered, medium, and high MIMAG) from CAMI challenge [2] are used for species and strain level dereplication evaluation.
Once representative MAGs are selected, the pipeline re-maps the sequencing data back to the MAGs and produces MAG-abundance profiles for all samples. The abundance profiles are used to compare the PGPB diversity between different samples. It can also be used to build co-occurrence networks involving assembled MAGs and known PGPBs.
The intermediate results of the EMGB pipeline are imported into the EMGB browser. In the browser, each individual sample can be selected and its computed results can be explored in a click-button style. Users can also compare different samples by selecting multiple samples in the browser tab. Selected metrics, such as the abundance tables of de-replicated MAGs from all metagenome samples, are also accessible through the web interface.
This project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 818431
(SIMBA). This output reflects only the author’s view and the Research Executive Agency (REA) cannot be held responsible for any use that may be made of the information contained therein.
References


[1] Huang et al. Analyzing large scale genomic data on the cloud with Sparkhit. Bioinformatics, 2018, DOI: 10.1093/bioinformatics/btx808.
[2] Sczyrba et al. Critical Assessment of Metagenome Interpretation—a benchmark of metagenomics software. Nature Methods, 2017, DOI: 10.1038/nmeth.4458.