Bielefeld Institute for Bioinformatics Infrastructure




Supporting local health authorities
in fighting the SARS-CoV-2 pandemic


Michael Beckstette; Bielefeld University, Bielefeld
The global SARS-CoV-2 pandemic poses numerous new challenges and actions on different kinds of administrative and institutional levels ranging from world-wide vaccination initiatives over state-specific changes of laws to new regulations for local authorities. Most notably with the appearance of new emerging virus variants like the more infectious Alpha (B.1.1.7) and Delta (B.1.617.2) variants, local health authorities in Germany were confronted with new tasks such as sequencing of virus genomes and virus lineage detection. The importance of these tasks emerged not only from the necessity to control local outbreaks but also from the obligation to provide data to federal government agencies like the
Robert Koch Institute (RKI) for monitoring of the pandemic situation and population risk assessment which is finally used to furnish recommendations to health professionals and governmental decision-makers.
“The researchers from BIBI were of great help in the bioinformatics analysis of our samples. They provided standardized and easy to use workflows, which have successfully been used to analyze and identify the variants of more than 260 SARS-CoV-2 positive samples.”
Dr. Henning Petersen from the CVUA-OWL
In early 2021, when the Alpha variant of the SARS-CoV-2 virus started to spread over Germany (Figure 1), the Chemical and Veterinary Investigation Office for the region Ostwestfalen-Lippe (CVUA-OWL) contacted researchers of the Bielefeld Institute for Bioinformatics Infrastructure (BIBI) and asked for support in the bioinformatics analysis of sequenced virus samples from patients from the OWL region.
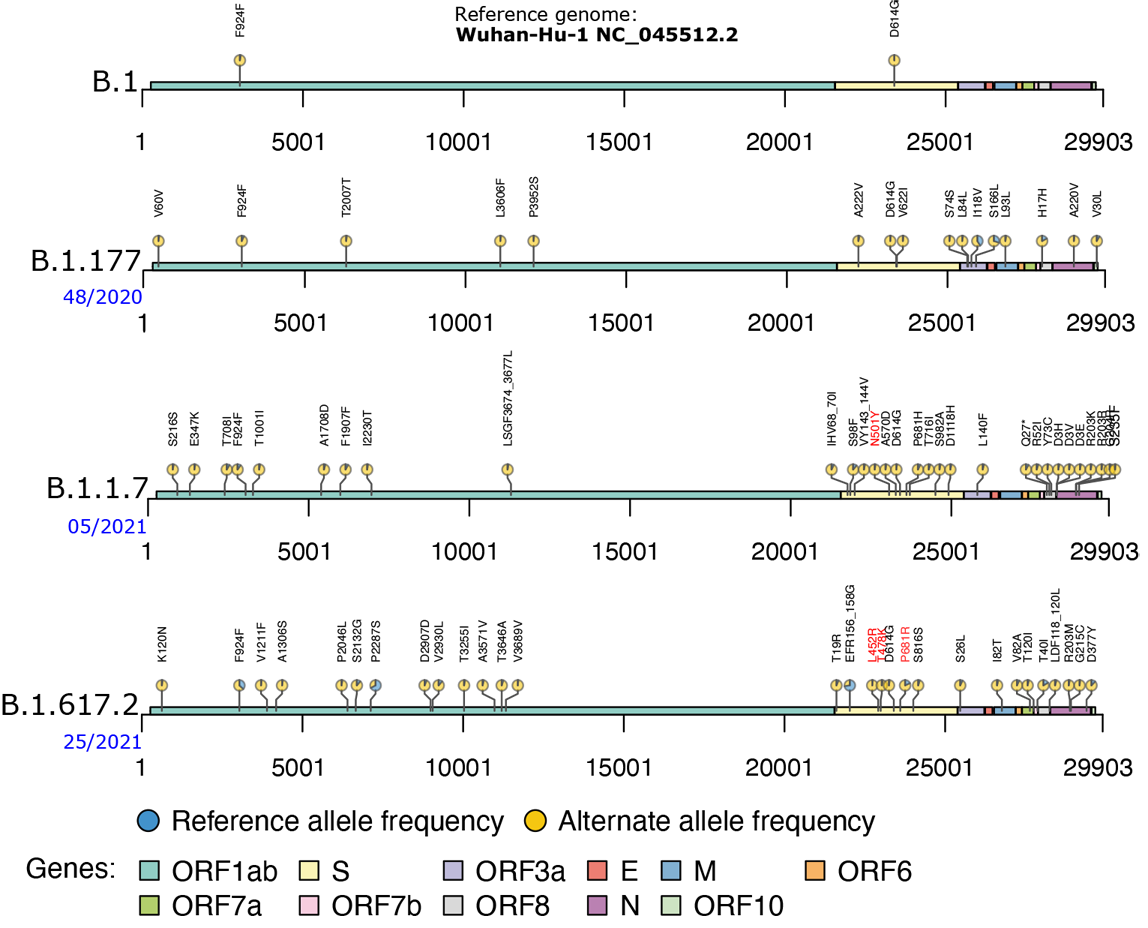
The genomic dynamics of SARS-CoV-2. Sketched are genomic variants – with respect to the original SARS-CoV-2 corona virus responsible for the initial outbreak in Wuhan/China – in protein coding regions of different virus strains circulating in Europe over the time of the pandemic. Variants are annotated with their allele frequencies and effect on the corresponding proteins amino acid sequence. If available, the calendar week of first occurrence in Ostwestfalen-Lippe (OWL), based on genome sequencing data of our project partner CVUA-OWL, is given in blue. The B.1 lineage is a large European lineage whose origin roughly corresponds to the Northern Italian outbreak in March 2020. It was the dominant lineage in Germany until summer 2020. The B.1.177 lineage spread mostly over Europa after opening of borders in summer 2020 and appeared for the first time in the OWL data in week 48/2020. B.1.1.7 (Alpha) is the first variant of concern (VOC) that spread over Europe. It was first detected in the United Kingdom in September 2020 and is associated with the N501Y mutation and with evidence for having higher transmissibility than other lineages resulting in rapid growth in the UK and internationally. At the beginning of 2021 (week 05/2021 in OWL) Alpha starts to push away other virus variants and quickly became the dominant variant in spring 2021. The B.1.617.2 (Delta) lineage was first detected in October 2020 in India and classified as a VOC in Mai 2021. It has much increased transmissibility compared to Alpha and is linked to a significantly higher risk of severe COVID-19 disease progression and death. In OWL it appeared in week 25/2021 and since July 2021 it has become the dominant virus lineage accounting today for more than 99 percent of all SARS-CoV-2 infections in Germany. With P681R, L452R and T478K it carries several mutations in the virus' spike protein (S gene) that are linked to higher virus load due to increased replication rates and the ability to partially escape neutralizing antibodies generated by the hosts immune response.
BIBI's substantiated existing expertise in this field [1, 2] and a pragmatic and efficient collaboration style between researchers from both institutions allowed to promptly provide a secure and easy to use solution operating on the federated de.NBI cloud computing infrastructure [3]. With the COG-UK [4] and the RKI CovPipe [5] analysis pipelines two widely used, fully automated bioinformatics workflows for the reproducible analysis of SARS-CoV-2 samples could be offered to CVUA-OWL researchers. The former has widely been used in the COVID-19 Genomics UK Consortium (COG-UK) project which has been a pioneering initiative in the use of large-scale, whole genome sequencing of SARS-CoV-2, with the aim to aid the harmonization of the analysis of sequencing data by providing a standardized analysis workflow. The latter is an alternative bioinformatics pipeline developed by the German Robert Koch institute (RKI) and widely used in Germany for the analysis of SARS-CoV-2 samples from viral outbreaks. Likewise, to the COG-UK pipeline, the workflow covers and automates all necessary steps from rigorous quality assessment of the input data, read mapping against the SARS-CoV-2 reference genome, over variant calling and generation of a consensus sequence of the virus containing the in the analysis process detected mutations (genomic variants) to lineage assignment. In addition, BIBI researchers enhanced these standard workflows with additional capabilities for comprehensive result visualizations (Figures 1 and 2) allowing to generate epidemiological information that is easily interpretable by public health institutions.
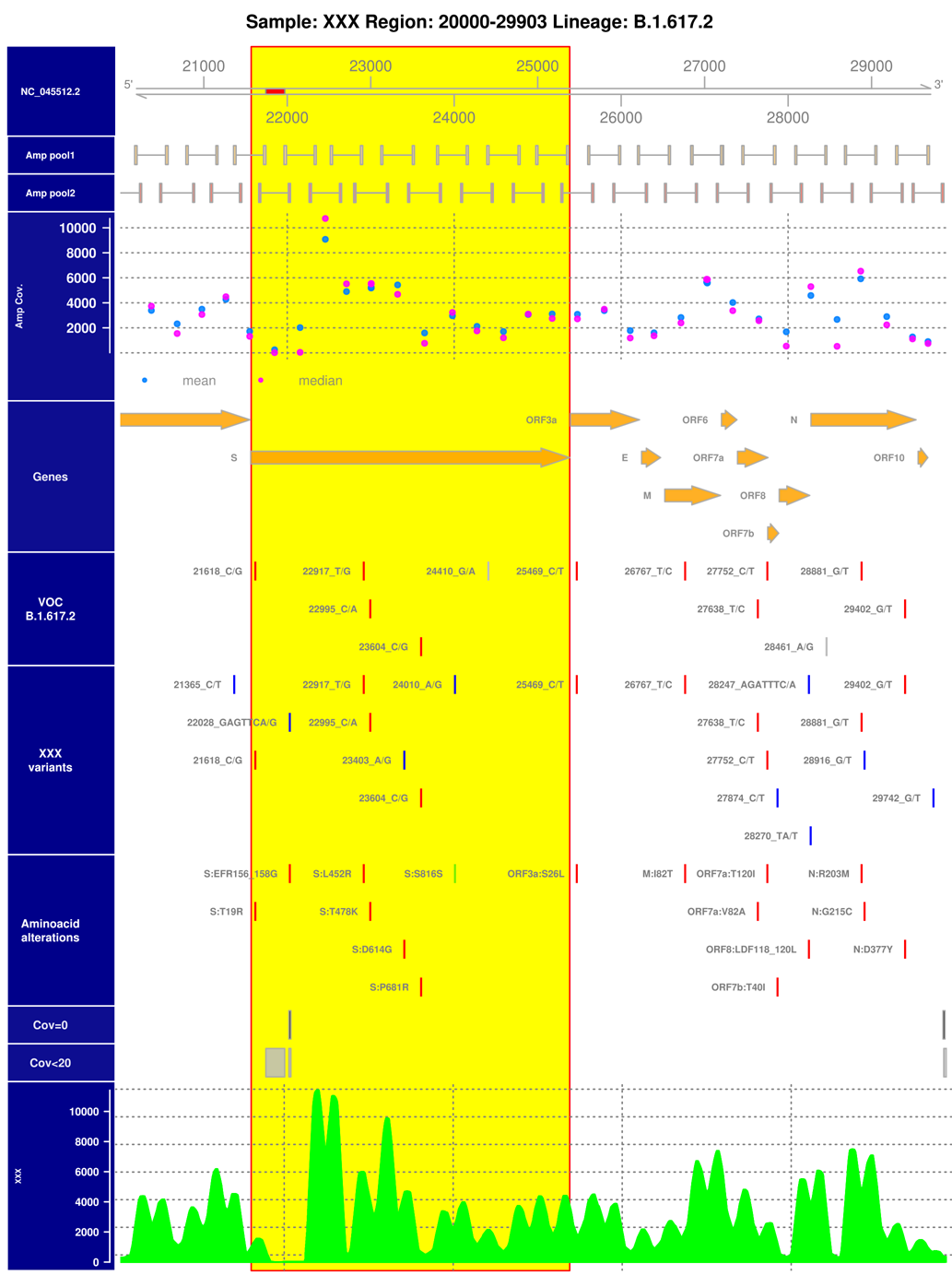
Analysis report. Analysis results of a SARS-CoV-2 positive sample from the Ostwestfalen-Lippe region that was classified as belonging to the highly infectious Delta variant (B.1.617.2) of the virus. Shown is an excerpt of the virus genome with coverage information of the used sequencing amplicons, gene annotation, VOC B.1.617.2 defining variants, variants called for this sample with their amino acid alterations and genomic coverage information. The part of the genome coding for the spike protein (S gene) is marked in yellow. Detected, lineage (Delta) specific genomic variants are colored red in the samples variant track.
References


[1] Schulte-Schrepping et al. Severe COVID-19 Is Marked by a Dysregulated Myeloid Cell Compartment. Cell, 2020. DOI: 10.1016/j.cell.2020.08.001.
[2] Brandt et al. Multiple Occurrences of a 168-Nucleotide Deletion in SARS-CoV-2 ORF8, Unnoticed by Standard Amplicon Sequencing and Variant Calling Pipelines. Viruses, 2021. DOI: 10.3390/v13091870.
[3] German Network for Bioinformatics Infrastructure – de.NBI: https://www.denbi.de/cloud
[4] COG-UK (ncov2019-artic-nf) pipeline: https://github.com/connor-lab/ncov2019-artic-nf
[5] RKI CovPipe pipeline: https://gitlab.com/RKIBioinformaticsPipelines/ncov_minipipe