Bielefeld Institute for Bioinformatics Infrastructure




Unlocking the genetic secrets of microorganisms
Accessing and analysing microbial genome data
In the last decade bioinformatics has silently filled in the role of cost effective and target-oriented data analysis. It has enhanced our understandings about the microorganisms' genome structure and the cellular processes in order to treat and control microbial cells as factories.
The genetic diversity of viruses on a graphical map


Alexander Schönhuth; Bielefeld University, Bielefeld
Various life-threatening viruses mutate insanely fast, thereby protecting the virus from human immune response or medical treatment. Naturally, virus variants form within the infected hosts, when hijacking the host’s replication machinery. Therefore, accurate tracking of strains within individual patients or local samples, for example obtained from wastewater, can make a crucial contribution to assessing the evolutionary course of epidemics [1].
We focus on developing methodology for identifying the development of new strains/variants, and to put them into context with existing strains/variants. To do that, all strains and variants are arranged in a “map-like” graphical data structure. This “map of variants” highlights the origin of new variants conveniently, and puts them into context with existing variants.
Further, this enables us to accurately place new infections on this map of variants. As a consequence, new infections can be classified at high resolution. Novel, emerging variants can be spotted quickly and integrated into the map in an evolutionarily consistent manner.
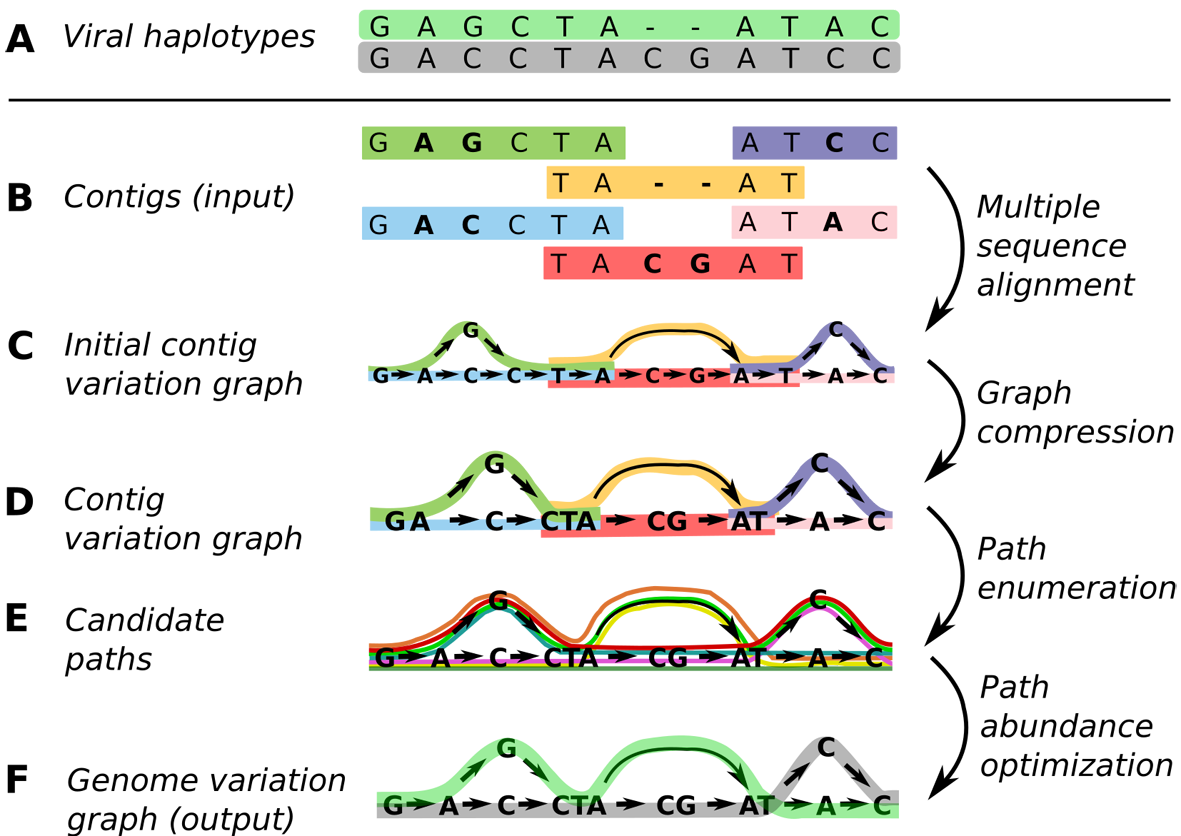
Pangenome graph construction
(A) Original viral haplotypes, reflecting strain specific genomes. While originally unknown, they are the source of fragments (aka contigs) shown in (B). The task is to reconstruct sequences shown in (A) from fragments shown in (B). This reconstruction proceeds in four further steps: computing a multiple sequence alignment leads to a first graph as shown in (C), compressing the graph by joining letters leads to (D). Eventually, inspecting all possible paths (“candidate paths” in (E)) and evaluating their plausibility relative to the original fragment data leads to the final situation, shown in (F). The final graph enables us to correctly identify the haplotypes that were responsible for generating the data.
Key to success is to make use of “pangenome graphs” as a relatively new concept to arrange individual, mutually related genomes in an evolutionarily sensible way. Pangenome graphs have recently been emerging, and gradually replacing traditional ways of working with genomes.
Their advantages are their compactness – which can save petabytes of storage space – their intuitive representation, and their consistency in terms of the evolutionary relationships among the individual genomes.
Recently, we have pointed out ways to make stringent use of pangenome graphs for tracking and analyzing viruses. Therefore, it was important to realize that not only one, but possibly several strains can populate individual hosts. In fact, this is rather common because new strains and variants form within hosts, when the virus hijacks the host’s replication machinery; note that virus particles cannot mutate while circulating between hosts.
The crucial first step is to adapt analysis tools accordingly, and to make it possible to construct pangenome graphs that reflect the within-host diversity of a virus: not considering within-host diversity collapses different mutations, which falsifies one’s view on the evolutionary development of the virus. The challenge however is that considering within-host diversity requires approaches that are essentially novel [2,3].
Once this foundation has been laid, accurate pangenome graphs can be constructed, as we could demonstrate in a corresponding series of papers [4,5]. See Figure 1 for an illustration of the algorithmic steps to be taken towards successful construction of viral pangenome graphs.
References


[1] S. Posada-Cespedes et al. Recent advances in inferring viral diversity from high-throughput sequencing data. Virus research, 2017. DOI: 10.1016/j.virusres.2016.09.016.
[2] J. Baaijens et al. De novo assembly of viral quasispecies using overlap graphs. Genome Research, 2017. DOI: 10.1101/gr.215038.116.
[3] X. Luo et al. Strainline: full-length de novo viral haplotype reconstruction from noisy long reads. In revision at Genome Biology. DOI: 10.1101/2021.07.02.450893.
[4] J. Baaijens et al. Full-length de novo viral quasispecies assembly through variation graph construction. Bioinformatics, 2019. DOI: 10.1093/bioinformatics/btz443.
[5] J. Baaijens et al. Strain-aware assembly of genomes from mixed samples. RECOMB, 2020. DOI: 10.1101/645721.